


Introduction to Protein Modeling Projects
Welcome to the Protein Modeling Projects Page at NSUWorks. NSU Halmos College of Arts and Sciences faculty members, Dr. Arthur Sikora, Department of Chemistry and Physics and Dr. Emily F. Schmitt Lavin, Department of Biological Sciences created this site as a repository for research projects in the field of protein modeling.
This site documents research primarily conducted by students enrolled in the honors course, HONR 1010G: Introduction to Protein Modeling which began in Fall 2021. Sikora and Schmitt were inspired by the Connecting Researchers, Educators and Students (CREST Program https://crestresources.org/ ) with assistance from 3d Molecular Designs and the former Center for Biomolecular Modeling (CBM).
Previously to and including projects from the Introduction to Modeling Course, students presented results of their protein modeling projects through the annual meeting of the American Society of Biochemistry and Molecular Biology with abstracts available here:
2017
2018
- Developing a physical model of O-GlcNAc transferase (OGT) in complex with TAB1
- Constructing a 3-D Molecular Model to Highlight the Conversion of the Normal Protein PrPC into the Mutated PrPSC in a Prion Disease
2019
2020
2021
Projects in this series include the presentation of results from teams of student researchers investigating protein modeling tools to describe their molecular “stories” of interest. We invite you to enjoy reading the results of research into these molecular stories told through protein models.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
-

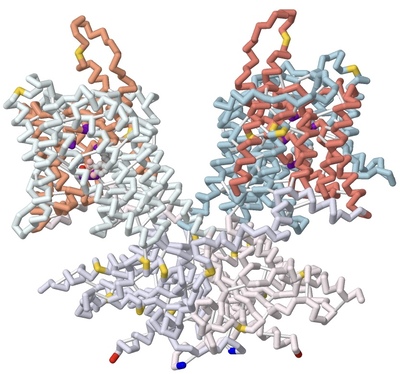
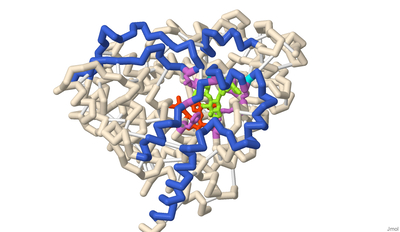
Using 3D Modeling to Describe the Electromotility of the Outer Hair Cell Protein Prestin, and its Role in Sound Perception Among Mammals
Marqus Colon, Syed Hussain, Russel Robert Reside, Emily Schmitt-Lavin, and Arthur Sikora
Prestin is one of the key motor proteins that has been identified in enabling auditory perception in mammals. By modulating its electromotility in response to changes in environmental voltage, prestin contracts and elongates in the plasma membrane of cochlear outer hair cells (OHCs). This allows different frequencies of sound to be processed quickly and precisely. Belonging to the SLC26A5 family of anion transporters, prestin is especially adept at binding anions in order to facilitate its oscillation through a series of unique conformations. Previous research has demonstrated that prestin is reversibly inhibited in the presence of salicylate. However, the broader mechanisms by which prestin senses and transduces voltage into cellular movement are not yet well understood. Previous studies have described the electromotility of prestin in terms of non-linear capacitance (NLC), wherein conformational changes in the protein are not linearly related to the voltage applied. High NLC is imperative for sound amplification in the cochlea, as this property enables OHCs’ selective response to different frequencies of incoming sound. 3D protein modeling was employed to better visualize the electromotility of this OHC protein by manipulating models of bottlenose dolphin (Tursiops truncatus) prestin available in the Protein Data Bank. Using the software PyMOL, Chain A of prestin in the inhibited state (7S9E) and Chain B of the compact, sensor-up state (7S8X) were spliced together into a novel merged model that depicts the fluctuation in the cross-sectional area of the transmembrane regions. This was possible because prestin is a protein homodimer whose peptide subunits could be swapped and replaced accordingly upon manipulation in the program. Key elements of prestin’s topology were then highlighted on this nascent model to emphasize the locations of the 14 gate and core transmembrane (TM) helices, the site of anion binding, and the cytosolic STAS domain. The colors corresponding with these regions include salmon, light blue, dark violet, and lavender, respectively. Helices TM3 and TM10 play a pivotal role in facilitating the movement of prestin’s dimers when bound to specific ligands. Likewise, the pocket formed by residues Gln97, Phe101, Phe137, Leu397, Ser398, and Arg399 has been identified as the anion binding site in the homodimer. The binding stability of ligands is further enhanced via additional noncovalent forces present in the active site, including pi stacking between salicylate and Phe137, and Ser393’s participation in hydrogen bonding. Arg399, the only positively charged residue in the cavity, is known to rotate up and down while the protein is moving, and neutralization of this key residue has also been found to eliminate prestin’s NLC entirely in vitro. Other residues of note that have been described by researchers include a series of 13 amino acid replacements (depicted in gold) that appear to be shared by several echolocating mammals, indicating convergent evolution between bats, whales, and dolphins.
-

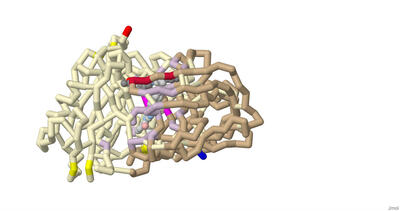
Modeling the Binding of Tetrodotoxin and Saxitoxin to the Nav1.7 Voltage-Gated Sodium Channel
Isabella G. Fiore, Smrithi Mukund, and Laasya Buddharaju
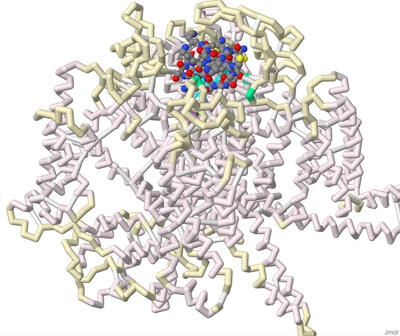
Approximately 1.5 billion people suffer from chronic pain, with 68 million people suffering in the U.S alone. Chronic pain is defined as pain that persists longer than 12 weeks despite medication or treatment. Within the U.S, about 5.5 million cancer patients experience chronic pain. Around 20% of these cancer pains have neuropathic origins and develop from radiation therapy. Halneuron, a new drug currently in development, seeks to provide pain relief to those suffering from chemotherapy-induced neuropathic pain. This drug includes Tetrodotoxin (TTX), a potent neurotoxin found in most species of pufferfish, as its main active ingredient. Our 3D printed model displays the structural details of Tetrodotoxin (TTX) as a pore blocker within the Nav1.7 channel, providing insight into its use as an analgesic. The model also shows the structural differences between TTX and a similar toxin, saxitoxin (STX), which has a lower binding affinity. A toxin produced by algae, STX is most concentrated in mussels and other shellfish that can induce paralytic shellfish poisoning. Both TTX and STX are Nav1.7 channel pore blockers that inhibit pain. TTX and STX were chosen from the Protein Data Bank and related literature studying pain inhibition. The PDB files 6J8J and 6J8H were used to develop a 3D molecular model showing how both toxins bind to the channel and inhibit action potentials. The files were imported to Jmol, where interactions and amino acids were highlighted to display binding differences. Our model contains key features such as the alpha subunit, backbone, and alpha helices of the Nav1.7 channel. The N and C terminals are intracellular, while voltage sensing domains are extracellular. The helix 4 (S4) of each of the voltage sensing domains have their positively charged lysine or arginine residues highlighted. Amino acids in the Nav1.7 receptor that bind to both STX and TTX are shown: Tyr362, Glu364, Arg922, Glu927, Glu930, Thr1409. Amino acids in the Nav1.7 receptor that bind to TTX, but not STX, are highlighted on the backbone: Gly1407, Gly1699, Phe1405, Ala1698. Amino acids in the Nav1.7 receptor that bind to STX but not TTX are highlighted on the backbone: Trp1700, Asp1744. The DEKA motif (Asp361, Glu930, Lys1406, and Ala1698) is marked on the backbone. Lys 1406 is the critical determinant that specifies the selective permeability of sodium over potassium in these voltage gated channels. The linker in between segments III-IV contains the fast inactivation motif (Ile1472, Phe 1473, Met 1471). There are 4 hydrophobic residues of note on the locus of the S6 of VSD IV: Leu398, Leu964, Ile1453, and Tyr1755. The tyrosine forms a down confirmation allowing sodium ions to be pushed through the channel. Overall, constructing this model allowed us to understand and visualize the binding differences between TTX and STX to the Nav1.7 channel, and simultaneously obtain insight regarding future applications in the treatment of pain disorders.
-

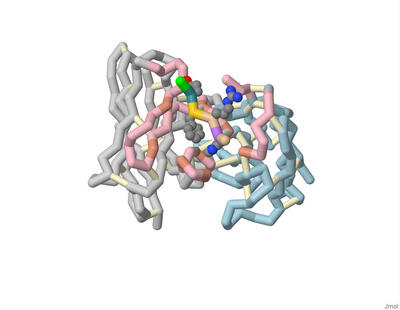
Modeling Cysteinyl Leukotriene Receptor Antagonist KNW for Possible Optimized Asthma Treatment
Shalet James, Sreejani Jonnalagadda, Emily Schmitt Lavin, and Arthur Sikora
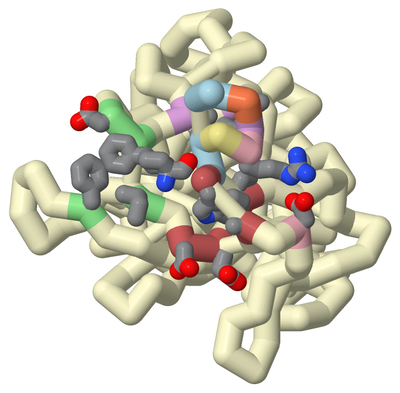
Underdiagnosed and under-treated, particularly in low- and middle-income countries, asthma has affected 262 million people globally. Current anti-asthmatic medications such as pranlukast inhibit cysteinyl leukotriene receptor 1 (CysLT1R), yet many patients do not respond to this drug. CysLT1R is associated with bronchoconstriction, inflammation, and mucus production in the airways of the lungs and bronchial tissues. When cysteinyl leukotrienes bind to CysLT1R, these effects are triggered contributing to the symptoms of asthma. The potential role of the related receptor CysLT2 in asthma remains poorly understood. To better understand this process, CysLT2R has been identified as a promising drug target for not only asthma but also other conditions such as brain injury and cancer. Students of the Honors Protein Modeling course at Nova Southeastern University modeled the interaction between a dual antagonist of CysLT1R and CysLT2R, KNW. A 3D model of KNW in complex with CysLT2R was based on PDB ID 6RZ6, modified using JMol, and 3D printed to showcase key interactions between drug and receptor. In this model of CysLT2R, we highlighted the ligand binding pocket, helix 8 (H8), and mutation residue interactions. The antagonist forms crucial interactions within the ligand-binding pocket (cyan). The N-linked carboxypropyl moiety forms salt bridges with Lys 37 and His 284 specific to CysLT2R (cpk). Mutating these residues to their CysLT1R counterparts decreases inhibition by antagonists. The key anchoring residue Tyr 119 interacts with benzoxazine, carboxylic groups, and amide linkers of the ligand (violet). The cleft opening residues include Leu 165, Val 208, and Tyr127 (light cyan). Unlike its counterpart, CysLT2R exhibits a wider cleft opening to the lipid membrane, enhancing ligand selectivity. Helix 8, a unique and flexible alpha-helix on the cytoplasmic side of the cell membrane, plays an important role in the regulation of G-protein activation and subsequent intracellular signaling cascades (pink). H8 conformation affects the binding site accessibility, signaling pathways, and receptor stability. Specifically, the salt bridge with Glu 310 stabilizes the junction between H8 and TM7 and the inactive state of the receptor when bound with the antagonist (cpk). Notably, the atopic asthma-associated mutation Met to Val in position 201 of CysLT2R results in a mildly impaired hypomorphic protein, with reduced ligand binding and inositol phosphate (IP) production (plum). Based on previously reported structure-activity relationship analysis, we developed a novel molecular inhibitor, Finlukast, aimed to have high affinity to both classes of receptors. Using SwissDock, we determined that this novel inhibitor molecule has high affinity binding to CysLT1 and CysLT2 receptors. Through the exploration and modeling of KNW, we gained further insight into the key structural interactions of dual antagonist KNW for similar receptor targets responsible for mediating the inflammation and bronchoconstrictive effects of cysteinyl leukotrienes.
-

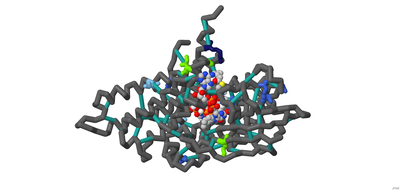
Modeling Calcium Binding to Yersinia pestis Type III Secretion Needle
Kaya Olszewski, Priyanka Prasanna, Chetana Movva, Arthur Sikora, Emily Schmitt Lavin, and Julie Torruellas Garcia
A type III secretion system (T3SS) is utilized by a variety of bacteria to inject toxins into host cells to cause disease, including Yersinia pestis, the bacterium that causes the bubonic plague. The Y. pestis T3SS needle subunit is a polymer made up of YscF proteins. Calcium is thought to play a role in regulation of toxin secretion by the YscF needle. High levels of Ca2+ binding inhibit secretion while low levels of Ca2+ binding cause secretion to be stimulated. According to a study done in 2005 by Torruellas et. al, different mutations of YscF can interfere with secretion regulation, most likely due to altered calcium interactions with the needle. Some details regarding which mutations lead to constitutive secretion (CS), no secretion (NS), or regulated secretion (RS) of the Yersinia outer protein (Yop) toxin are known. The goal of our research is to understand how the mutations present in previous wet lab experiments affect calcium binding to the YscF hexomer subunit. To understand if and how calcium interacts with the YscF needle to inhibit Yop secretion, an accurate model representation of the needle-calcium interactions is required. To model this phenomenon, the interactive accelerated protein prediction tool, CollabFold, was used to create probable hexamer subunits of the needle with the mutations from the previous experiment (Torruellas et. al, 2005). MIB2, a binding prediction modeling server, was used to predict binding with Ca2+, Fe2+, Fe3+, Zn2+, Cu2+, Mg2+. Alternative divalent cations were used to compare to Ca2+ binding to the hexamers. Ca2+ bound to the wild type (WT) hexamer at 12D, 15D, 16L; 28D, 29D; 36D, 37A with an average binding potential of 2.4. The binding sites contain aspartic acids that lead to CS when mutated to alanine, suggesting those binding sites play a role in calcium regulation of the needle. Zn2+ and Fe2+ bound to the WT hexamer at 56, 60 with an average binding potential of 4.0. Zn2+ and Fe2+ bound with higher binding potentials to the WT mutant hexamers on average compared to Ca2+. The hexamer structures of the different mutations were compared to look for structural differences between the mutations. Arginine 73 creates a ring in the center of the WT hexamer. The arginines at position 73 are further apart compared to the WT hexamer in the double mutant (D28A, D46A), I13A, and D17A CS hexamer mutations. The arginines at position 73 were obstructing the center passage in the N31A and D77A NS hexamer mutation. The arginine ring structural comparison between hexamers suggests that the R73 ring may play a role in regulating Yop secretion by obstructing the central needle passage. A 3-d printed model was created of a possible hexamer of the YscF Y. pestis needle subunit with the residue mutations causing constitutive secretion of Yop, and corresponding mutations indicating no Yop secretion, each highlighted in designated colors. The mutations indicating constitutive secretion are I13A, D17A, D28A, D46A. The mutations leading to no secretion are N31A, V34A, D77A, D77C, I82A, I82C. The results determined what mutations lead to CS, NS, or RS of the Yop toxin in the presence or absence of calcium.
Torruellas, J., Jackson, M., Pennock, J., Plano, G. (2005). The Yersinia pestis type III secretion needle plays a role in the regulation of Yop secretion. Molecular Microbiology, Vol 57(6), 1719-1733, https://doi.org/10.1111/j.1365-2958.2005.04790.x
-

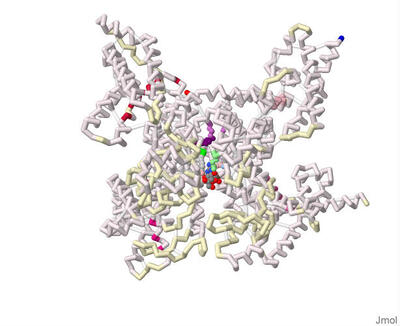
Identifying the Binding Residues on CYP3A4 to Naringin using Protein Modeling and Docking
Parth Shah, Vraj Patel, Sanjana Ananthula, Emily Schmitt Lavin, and Arthur Sikora
Cytochrome (CYP) enzymes are a superfamily of monooxygenase hemoprotein enzymes that are found throughout the body but are heavily concentrated in the endoplasmic reticulum and mitochondria of liver cells. These enzymes catalyze reactions that modify a wide range of substrates into more hydrophilic and, therefore, more readily excreted forms. Cytochrome enzymes are heavily involved in the detoxification process of many medically relevant drugs. As such, the inhibition and activation of these enzymes can substantially alter the effective bioavailability of medications and can introduce additional variables or modifiable variables into a pharmacological-based treatment.
Cytochrome p450 3A4 (CYP3A4) is one of the most abundant cytochrome enzymes and can target a wide range of substrates, including many medically relevant drugs. Inhibition of CYP3A4 can increase the bioavailability and duration of the availability of a medication in the bloodstream. This makes the factors associated with the inhibition and activation of CYP3A4 of great medical interest and importance. CYP3A4 has been noted to be inhibited by naringin, a flavanone found in grapefruits and other citrus fruits. However, the characteristics of naringin binding to CYP3A4 are unknown.
The residues at which naringin binds to CYP3A4 were identified using 3-D protein modeling and computerized molecular docking simulations. The PDB file for the CYP3A4 enzyme (8DYC) was obtained from the RCSB Protein Data Bank and was manipulated to remove the substrates, leaving only the prosthetic heme group. The binding between CYP3A4 and different inhibitors was studied in the literature to identify specific features of the CYP3A4 protein. The residues that form the opening of the protein cleft, the area where the substrates bind and the residues that form the active site of the enzyme were noted for reference for the docked position of naringin. PyRx was used to conduct molecular docking simulations to predict the location of naringin binding in the CYP3A4 structure. However, due to computational limitations, the docking did not predict interactions with the HEME group. Rather the final docked structure of naringin to the CYP3A4 enzyme was produced using the coordinates of the docked naringin and the HEME-containing structure of CYP3A4. The Contacts/Clashes functionality in USCF Chimera was used to identify the specific residues on the surface of the protein and these residues were compared to those identified through the literature consulted.
The residues of CYP3A4 that are involved in binding naringin that were identified through this process were: Glu 374, Arg 372, Arg 106, Arg 105, Ala 370, Phe 215, Arg 212, Phe 304, Leu 482, Ser 119, Ile 223, Thr 224. Of these, Arg 372 and Thr 224 were also noted in the literature to be involved in forming the opening of the cavity in the protein. The residues Glu 374, Arg 106, Arg 105, Ala 370, Phe 215, Arg 212, Phe 304, and Ser 119 were also noted in the literature and are likely involved in forming the active site. The residues Ile 223 and Leu 482 were not noted in the works consulted and may represent additional or potentially novel residues that may be involved in the inhibition of CYP3A4. Overall, the results of the molecular docking suggest that naringin inhibits CYP3A4 by binding and blocking both the opening of the cleft within which the substrate binds and binding to residues in the active site of CYP3A4.
-
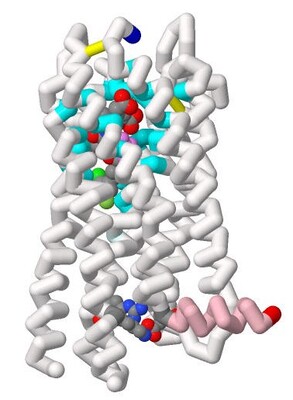
 Inhibitor Aminoquinoline (68K) for Possible Treatment of Alzheimer's Disease by Juhi Dalal, Shreya Averineni, Pranav Madadi, Emily S. Lavin, and Arthur Sikora")
Binding of Beta-site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE1) Inhibitor Aminoquinoline (68K) for Possible Treatment of Alzheimer's Disease
Juhi Dalal, Shreya Averineni, Pranav Madadi, Emily S. Lavin, and Arthur Sikora
Alzheimer’s Disease (AD), affecting approximately 24 million people worldwide, is characterized by the formation of amyloid-β plaques within the brain. Alzheimer’s research has been focused on limiting amyloid-β production through developing inhibitors for the enzymes needed within the amyloid cascade. This project focuses on the aminoquinoline class of inhibitors, of which 68K (PDB: 5i3Y) is the most effective because of its strong Kd and IC50 values. The students of the Honors Protein Modeling class at Nova Southeastern University modeled the interaction between Beta-site Amyloid Precursor Protein Cleaving Enzyme 1 (BACE-1) and 68K. Using Jmol a model was developed, and 3D printed to show how the inhibitor (68K) fit into the enzyme’s active site. This model highlights important aspects of the interactions between the ligand and the BACE-1 enzyme. 68K has strong interactions with 32 amino acid residues in BACE1, some of which are intertwined with one another. For example, BACE-1’s residues Val69, Pro70, and Tyr71 are known collectively as “the flap”. “The flap” is a β-hairpin loop structure that is positioned directly over BACE-1’s catalytic dyad, a group of amino acids within the active site of the enzyme. “The flap” is also responsible for regulating access to the enzyme’s catalytic dyad (Asp 32 and Asp 228) by a given substrate (or inhibitor). Researchers found the inhibitor 68K to have interactions with the flap which maximizes the strength of the interaction with BACE-1 residues, thus minimizing the distance between the inhibitor’s various functional groups and accommodating their specific polarities. Being able to visualize the protein structure using a 3D model aids in the understanding of how the ligand inhibits this enzyme leading to the progression of AD.
-
 on Reducing Amyloid-Beta Plaques by Nikhila Paleati, Pranav R. Neravetla, Akhil B. Godbole, Emily S. Lavin, and Arthur K. Sikora")
Comparing Effectiveness of Two Antibodies (Aducanumab and Gantenerumab) on Reducing Amyloid-Beta Plaques
Nikhila Paleati, Pranav R. Neravetla, Akhil B. Godbole, Emily S. Lavin, and Arthur K. Sikora
Alzheimer’s disease (AD) is a degenerative neurological disorder that destroys memory and other important cognitive functions. As time progresses, brain cell connections, as well as the brain cells themselves, atrophy and die. AD is caused by a missense mutation in the amyloid-beta peptide within the amyloid precursor protein (APP). The mutation results in glutamine being replaced with glutamic acid. Previously conducted studies showed that mutated forms of the amyloid-beta peptide fragment have a greater tendency to stick together and form protein clumps or aggregates. The abnormal build-up of aggregates in and around the brain cells has been found to be strongly associated with the development of Alzheimer’s disease, therefore, it appeared crucial to study the methods that reduce these build-ups.
Attempts to treat this disease have produced antibodies that bind to the mutated amyloid-beta peptide and clear the aggregated amyloid precursor protein out of the brain. The overall goal of this project is to use 3D printed protein models to show interactions leading to a clearer explanation of the efficacy variations between antibodies. One antibody, Aducanumab, is currently in Phase 3 clinical trials and has been fast-tracked by the U.S. Food and Drug Administration. Aducanumab functions by specifically binding to the mutated amyloid-beta peptide and clearing aggregates out of the brain. This antibody binds to a smaller linear epitope formed by amino acids 3-7 of the amyloid-beta peptide. Using Jmol, protein visualization software, the Aducanumab (6CO3) PDB was manipulated to highlight multiple hydrophobic interactions, shown in a dark salmon color, and 2 hydrogen bonds, shown in white. The small binding location, flexibility provided by fewer strong interactions, and high affinity for aggregates at a high density make the antibody ideal for clearing out large aggregates.
Another antibody, Gantenerumab, is still undergoing testing in order to ensure safety and efficacy. This antibody functions by binding to a longer linear epitope formed by amino acids 3-11 of the amyloid-beta peptide. Unlike Aducanumab, Gantenerumab interacts with peptides through 2 salt bridges in addition to 3 hydrogen bonds and multiple hydrophobic interactions. Along with hydrogen bonds in white and hydrophobic interactions in dark salmon, the Gantenerumab (5CSZ) PDB was manipulated to show negative side chains of the salt bridge, labeled in red, while the positive side chains were labeled in blue. The increased number and strength of interactions reduces the flexibility of this antibody, thus making it difficult to easily bind and clear aggregated peptides. While both antibodies bind to a similar region of the amyloid-beta peptide and function to remove aggregates, they vary in the amount and type of interactions made with the amyloid-beta peptide.
-
 by Omar E. Saleh, Rhea Khatiwala, and Jeremy Ignatius")
Investigating The Mechanisms of Active Site Mutations to the 1T9G WT MCAD Protein to Better Understand Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCADD)
Omar E. Saleh, Rhea Khatiwala, and Jeremy Ignatius
Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCADD) is a human disorder that hinders β-oxidation, affecting approximately 1 in 17,000 people in the United States. Once mutated, the Acyl-CoA Dehydrogenase Medium-Chain (ACADM) gene, which is solely responsible for MCADD, is unable to produce enough MCAD enzymes to metabolize medium-chain fatty acids. As a result, fats are not catabolized, causing symptoms of lethargy and hypoglycemia, as well as damage to the brain and liver due to a buildup of unused fatty tissue. The purpose of this project was to investigate the possible and known effects of different amino acid mutations on the human MCAD protein and produce a 3D-printed model to explain the molecular story of MCADD. This model builds on previous bioinformatics and in vivo experiments aimed at revealing the underlying enzymatic mechanisms of MCADD. Using PyMOL, the human wild-type MCAD (PDB ID: 1T9G) had its electron transferring flavoprotein (ETF) complex removed and a single chain from its homotetramer portion isolated for clarity. PyRx was used to dock the substrate, Octanoyl-CoA (PDB ID: CO8) into the slightly mutated enzyme, referencing PDB ID 1EGC. Known mutations from the PDB files and related literature were then compared and analyzed on the modified 1T9G to determine known and possible effects the mutations had, such as helix-helix stability and ligand hydrogen bonding. LigPlot+ was then used to analyze ligand-active site interactions. Jmol was used to cosmetically enhance the modified 1T9G to produce a 3D model for printing. In the model, the mutations were ranked according to known KM range values (0.4, 0.6), (0.7, 0.9), and 0.9+, which were highlighted in the colors “lightskyblue”, “royalblue”, and “midnightblue”, respectively; unknown KM values were colored “chartreuse”. All mutations had their side chains shown for further clarity. E376, the catalytic base, was colored “magenta”, the backbone was colored “dimgray”, and the support struts of the model were colored “lightseagreen”. The use of the 3D model was beneficial, enabling model viewers to locate, determine, and hypothesize the mutations and their effects on MCAD, in addition to providing a visual and physical learning aid for researchers, professors, students, and other biomedical professionals. Furthermore, the clarity produced by a physical model ultimately enables further research for MCADD and may assist in the development of a cure for those who unfortunately suffer from this rare condition.
-

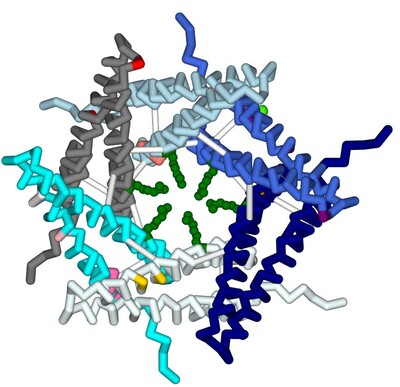
Modeling the binding of ω-conotoxin and other toxins to the N-type voltage-gated calcium channel
Serena Sha, Sophie Welch, and Ashley Guillen-Tapia
Approximately 1.5 billion people in the world suffer from chronic pain, persistent pain that carries on for longer than 12 weeks despite medication or treatment. Management of chronic pain typically includes the use of non-steroidal anti-inflammatory drugs (NSAIDs) or prescription pain medications, including opioids. An alternative therapy derived from conotoxins, toxins released from marine predatory snails in the family Conidae, was approved for the treatment of severe chronic pain in 2004. This pharmaceutical has the trade name Prialt and is also known as ziconotide. Once ziconotide is in the human body, it acts as a channel blocker of the N-type voltage-gated calcium channels, also known as Cav2.2. The literature and related PDB files were manipulated using PyMol and Jmol to create a 3D-printed model to explain the molecular story behind how a particular conotoxin binds to a calcium-gated ion channel. Additionally, computer visualization tools were used to show how several related toxins from other organisms would be expected to dock to the calcium ion channel. The 3D-printed model highlights specific features that contribute to the ω-conotoxin (MVIIA) binding to the calcium channel alpha 1B subunit as described in the literature (PDB: 7MIX). These conotoxins have a very characteristic disulfide bond linkage pattern which plays a role in the correct folding of the peptide and stabilization of its structure. In MVIIA, the non-cysteine amino acids form unstructured loops affecting binding affinity and calcium channel-blocking activity. Of particular interest is the second loop located between Cys8 and Cys-15. It appears to be exceptionally important in directing selectivity toward N-type calcium channels and away from P/Q-type calcium channels. Ziconotide does not directly seal the entrance to the vestibule of the selectivity filter, but it blocks ion entrance by neutralizing the outer electronegativity and sterically hindering the ion access path to the entrance of the selectivity filter. Salt bridges are formed between Arg10 and Tyr13 on ziconotide and Asp664 of the channel. Four of the eight ziconotide-coordinating residues, Thr643, Asp1345, Lys1372, and Asp1629 in Cav2.2 are not conserved in other calcium channels which may explain the subtype specificity of pore blockage by ziconotide. The EEEE motif consisting of Glu314, Glu663, Glu1365, and Glu1655, determines the Ca2+ selectivity. Also included in the model are the receptor's alpha helices and bound calcium ion. The N terminus and C terminus of the receptor are labeled in blue and red respectively to orient the model.
No crystal structures are available for ω-conotoxins bound to several other types of N-type calcium channels. To investigate the potential calcium channel blocking properties of conotoxins MVIIC, GVIA, MoVIB (from the cone snail, Conus magus) and ω-agatoxin IVA (from the spider, Agelenopsis aperta), the computer-based tool ROSIE was used to simulate binding of these peptides to the Cav2.2 channel. As expected, the toxin shown in the crystal structure of 7MIX, bound best to the Cav2.2 channel. The toxins MVIIC, GVIA, and MoVIB bound with lower affinity. The agatoxin IVA did not have any relevant binding to this calcium channel. Overall, protein modeling allowed for a deeper understanding of how conotoxins bind to and block the calcium channel possibly leading to additional therapeutic approaches to pain relief.
Support or Funding Information:
This work was made possible by funding through the National Science Foundation, Division of Undergraduate Education (NSF-DUE) grant number 1725940 for the CREST Project. Nova Southeastern University’s Farquhar Honors College and Dept. of Biological Sciences also provided support. Protein model printing was made possible by 3d Molecular Designs.
-

Exploring structural differences between antagonistic peptides for the development of orally bioavailable PCSK9 inhibitors
Bhavya Soni and Pritika Vemulapalli
Familial hypercholesterolemia (FH) is an autosomal genetic disease that causes elevated blood levels of low-density lipoprotein (LDL). One of the leading causes of FH is gain-of-function mutations in the gene coding for proprotein convertase subtilisin/kexin type 9 (PCSK9). The PCSK9 protein binds to LDL receptors (LDLR) on the surface of hepatocytes and promotes their degradation, preventing the recycling of LDLRs and thus increasing LDL blood levels. Monoclonal antibody therapies that bind to PCSK9 inhibiting LDLR binding are currently only available as an injection. However, several orally bioavailable PCSK9 inhibitors have been formulated and are undergoing clinical trials. One such therapy contains small-molecule-peptide inhibitors that bind to a cryptic site (N-terminal groove) adjacent to the LDLR binding site located in the catalytic domain. A helical region (S153-I161) is contained within this groove with conformational flexibility leaving the area open to small-molecule peptides. The peptide must consist of two components: a helical peptide with a high binding affinity to PCSK9 and an appended extension with antagonistic properties to inhibit LDLR binding. The extension must encroach upon the LDLR binding site’s hydrophobic pocket which significantly contributes to the binding energy of LDLR. Using two known PDB structures containing the removable peptides (A (5VLP) and B (6U3I)) in the N terminal grove, a three-dimensional printed model was created to demonstrate the interactions and proximity to the hydrophobic pocket. This model highlights critical amino acids on the peptides and PCSK9 to emphasize how the interactions support certain substitutions. One significant interaction lies between PCSK9 residues Ile369, Phe379, Asp238, and Ala239 and Peptide B’s organic moiety (1-amino-phenylcyclohexane-1-carbonyl). These PCSK9 residues surround the LDLR binding region's hydrophobic pocket indicating a successful inhibition. On the other hand, Peptide A’s FPG motif added to the Trp1 anchor formed a beta-turn which was unable to reach and, therefore, interact with PCSK9 residues Ile369, Phe379, Asp238, and Ala239. Through the 3D model, it was visualized that Peptide A had a beta-turn that precluded further extension into the target site, limiting its antagonistic ability. The model also highlighted Peptide B’s attached organic moiety which reaches the hydrophobic pocket in the proximal LDLR binding region, as shown in the literature to increase the binding affinity by >100 fold with reduced overall mass for an improved oral therapeutic.
-

How can we design an inhibitor with an enhanced binding affinity that is selective for MMP12 ?
Aisha Y. Abdool, Lyla Abbas, Tassnime Sebaei, Emily Schmitt, and Arthur Sikora
Matrix metalloproteinase 12 (MMP 12) is one of the twenty-three members of the peptidase M10 family which are primarily responsible for the breakdown of the extracellular matrix. MMP12 plays a key factor in the degradation of elastin and is commonly studied in the lungs of smokers, where MMP12 digests the elastin and serves as a chemokine to recruit a pro-inflammatory immune response. Thus, MMP12 is a major therapeutic target in wound healing and scar formation following a myocardial infarction. Students from the Honors Protein Modeling class at Nova Southeastern University modeled the interactions between MMP12 and various inhibitors. Using the protein data bank, an MMP12 protein complexed with the inhibitor called EEG under the code 3LIK was discovered. The structure was imported into JMOL: a protein visualization software. EEG intercalates into the S1 loop of the MMP12 protein without causing any disturbance to the loop's conformation. Murine trials were found with corresponding data for another MMP12 inhibitor known as AS111793 which was shown to reduce inflammation associated with cigarette smoke. A series of inhibitors were created using key components of EEG and AS111793. The binding was modeled on Py-Rx: a screening software used to dock the inhibitors. It was found that the hybrid compound created had a higher binding affinity than AS111793, but less affinity than EEG. This may be because a majority of the solvents and elements were removed from the inhibitor which did not allow the docking to occur.
-

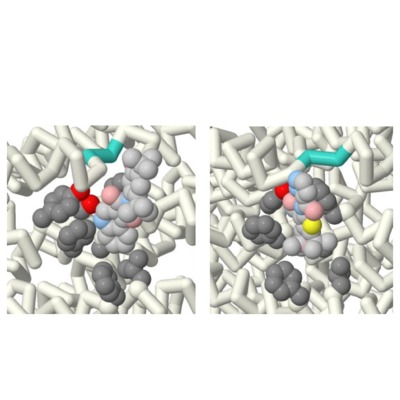
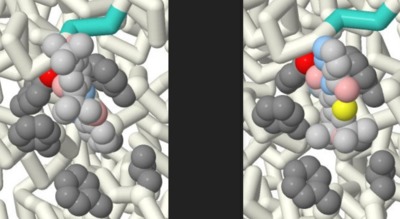
Comparing the Effectiveness of Ivacaftor and GLPG1837
Jordan Nichole Carreras, Saimi Reyes, Vibha Sankavaram, Emily Schmitt Lavin, and Arthur Sikora
Cystic Fibrosis is a fatal autosomal recessive genetic disease that effects more than 70,000 people worldwide. This disease is caused by a mutation within the CFTR causing an excess of mucus within the lungs, and effects other cells that produce bodily fluids (i.e. sweat and digestive fluids). The aim of this project was to identify the key differences between two similar potentiator drugs, Ivacaftor (Kalydeco®) and GLPG1837, that are used in the treatment of Cystic Fibrosis. The research question posed was: How does a model show the difference in efficacy of GLPG1837 compared to Ivacaftor? The comparison was done through the research and rendering of protein models through the Jmol software. The Jmol files of the drugs were taken from the Protein Data Bank (PDB). The files that correlate to the drugs are 6O2P (Ivacaftor) and 6O1V (GLPG1837). The files displayed the drugs within the CFTR channel attached to their binding sites. Additionally, further research was done on their respective chemical compositions through PubChem. The noticeable differences between the two drugs were their chemical composition, despite binding to the same active site within the CFTR. There is sulfur found within the hinge region (binding site) of the CFTR. Due to GLPG1837 containing a sulfur molecule and sulfur’s high binding affinity for itself, this appears to be the cause of GLPG1837’s increased efficacy over Ivacaftor. Due to the noticeable difference of the drugs displayed in the models it is concluded that an efficacy can be shown between the medications through modeling. Furthermore, it’s recommended that models such as these be used in the process of providers explaining treatments to patients who are using these medications. By providing tools to break down the science behind these medications to all age groups it can further rapport between patient and provider. As well as reduce stress of patients due to a better understanding of their treatment.
-

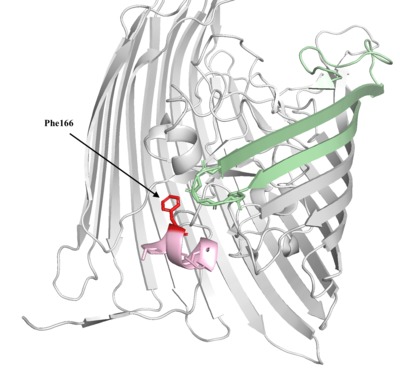
Limiting Iron Acquisition of E. Coli With Anti-TonB1 and AntiTonB2
Jose Diaz, Ryan Luib, and Seethal Doki
The bacteria E Coli infect approximately 200,000 individuals in the United States and can lead to critical illnesses. The transfer of iron from the bloodstream to bacteria is one of the main requirements for the survival. One of the mechanisms to obtain iron by use of siderophores and the protein FhuA was investigated. Undergraduate students grouped into teams to each explain a unique molecular story- modeled in Jmol/Pymol and demonstrated through a poster and PowerPoint. Using the PDB file, 2GRX, a drug to inhibit the TonB-Ton Box interaction which provides the FhuA iron mechanism energy to operate was researched. A protein model was designed to bind to the Ton Box region of FhuA with higher affinity by altering polar and charged residues to nonpolar or aliphatic residues that were present in the original TonB protein through programs Pymol and Jmol. The presence of many nonpolar amino acids in residues 8-16 and 588-592 of FhuA and the charged amino acids in residues 166-170 and 225-235 of TonB suggest that mutations of R166F, N227L, K231A on the tonB will lead to a protein with a stronger interaction with the Ton Box. The change from Arginine 166 to Phenlyalanine facilitates a nonpolar-nonpolar interaction between the TonB and Alanine of FhuA. Similarly, mutating polar asparagine to nonpolar leucine and the change of positively charged Lysine to uncharged Alanine will allow a stronger interaction. These hypothesized interactions of the mutated Anti-TonB1 with the FhuA are based on predicted outcomes because of the limitations of programmed docking of large proteins. Through the research and design of the course, students were able to develop skills with protein interface programs such as Pymol, Jmol, and Pyrx. This project was made possible by Nova Southeastern University and the guidance from Dr. Arthur Sikora and Dr. Emily Schmitt Lavin.
-

Highlighting How the Structure of Marine Bacterial Laminarinase Can Improve Biogeochemical Cycling During Global Climate Change
Heidi Hellenbrand, Rachel Harris, and Chino Villanueva
Marine bacterial laminarinase cuts polysaccharides in the process of remineralization, a key process in the ocean biogeochemical cycling of nutrients. We reviewed the protein model of 6JH5 in Jmol to highlight the important areas of the model that relate to thermostability and function and the effects of global climate change on the protein. The catalytic cleft of Glu135 and Glu140 was vital to the depolymerization mechanism. Substrate chain positions 130-143, specifically Trp130 being used for recognition, were important to the proficiency of the structure. The calcium ion on the opposite side of the β-sheet from catalytic cleft increased its degrading activity and thermostability. The residue interactions with Glc(−1) and Glc(−2) were unveiled to be crucial for β-1,3-glycosidic bond selectivity by the enzyme. Previous studies also showed the residue interactions were also important to the protein’s thermostability and thermophilicity. Our research found that the protein’s function will be negatively impacted by global climate changes as temperature beyond the protein’s optimal temperature (6JH5: 20°C) caused by climate change will also decline activity as hydrogen bonds between proteins weaken and being to denature. The function of our protein is important in the biogeochemical cycling of nutrients in the ocean and without its high activity, the cycling of nutrients and DOM like carbon and nitrogen won’t be as proficient.
-

Investigating the Structure of Potential New Drug to Treat Sickle Cell Anemia through Inhibition of the Polymerization of Hemoglobin S
Brianna M. Lacasse, Isadora R. De Abreu, and Rathika Manikandan
Sickle cell anemia is a hematologic disorder impacting over 15 million people worldwide. It is caused by a single point mutation in the gene hemoglobin-Betha, where a glu group is replaced by val (GAG --- GTG) in the seventh codon (glu7val) of chromosome 1. In this study, we are comparing the anti-sickling properties of drugs in varied conditions in order to create a drug that is effective in an O2-independent manner and with a 1:1 stoichiometry for lower dosage purposes. We used Pymol and Jmol to compare the structures of the aldehydes GBT-440 and VZHE-039, which interact on the same binding site to treat sickle cell disease. GBT-440’s bulkiness allows it to have a 1:1 stoichiometry, while VZHE-039’s solubility is due to its interaction with the hemoglobin’s alpha cleft, allowing it to be O2-independent. We identified the pyridine and pyrazole structure from GBT-440 and the methyl hydroxy moite from VZHE-039 as key structures, and created a hypothetical new drug, a hybrid of VZHE-039 and GBT-440. The pose predicted would allow the drug to interact with the sickled hemoglobin in a 1 to 1 ratio and in an O2-independent manner.
-

Comparing the Effectiveness between Ivacaftor and GLPG 1837
Saimi Vanessa Reyes, Jordan Nichole Carreras, and Vibha Sankavaram
Cystic fibrosis is an autosomal disease that is caused by a defect in the CFTR, or cystic fibrosis transmembrane conductance regulator. It affects more than 70,000 people world-wide and is life-threatening to those affected. To treat this disease there are two drug classes: correctors and potentiators. Correctors aim to correct the misfolded CFTR proteins, while potentiators aid in increasing ion concentration outside of the cells by keeping the channels open for longer periods of time. The aim of this project was to identify the key differences between two similar potentiator drugs, Ivacaftor (Kalydeco®) and GLPG 1837. The research question posed was: How does a model show the difference in efficacy of GLPG1837 compared to Ivacaftor? The comparison was done through the research and rendering of protein models through the Jmol software. The Jmol files that were used were 6O2P (Ivacaftor) and 6O1V (GLPG 1837) from the Protein Data Bank (PDB), which displayed the drugs in their binding site within the CFTR protein. It was shown that GLPG 1837 contained a sulfur molecule, while Ivacaftor did not. It is believed that this difference is what causes GLPG 1837 to appear more effective than Ivacaftor (in clinical trials) because the hinge region of the CFTR protein also contains a sulfur molecule. These models can be used by providers to explain treatments to patients who are using these medications much more effectively. By providing tools to break down the science behind these medications to all age groups it can further rapport between patient and provider. As well as reduce stress of patients due to a better understanding of their treatment.