


Files
Submission Date
Fall 2022
Abstract
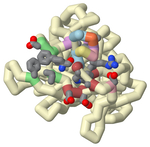
Familial hypercholesterolemia (FH) is an autosomal genetic disease that causes elevated blood levels of low-density lipoprotein (LDL). One of the leading causes of FH is gain-of-function mutations in the gene coding for proprotein convertase subtilisin/kexin type 9 (PCSK9). The PCSK9 protein binds to LDL receptors (LDLR) on the surface of hepatocytes and promotes their degradation, preventing the recycling of LDLRs and thus increasing LDL blood levels. Monoclonal antibody therapies that bind to PCSK9 inhibiting LDLR binding are currently only available as an injection. However, several orally bioavailable PCSK9 inhibitors have been formulated and are undergoing clinical trials. One such therapy contains small-molecule-peptide inhibitors that bind to a cryptic site (N-terminal groove) adjacent to the LDLR binding site located in the catalytic domain. A helical region (S153-I161) is contained within this groove with conformational flexibility leaving the area open to small-molecule peptides. The peptide must consist of two components: a helical peptide with a high binding affinity to PCSK9 and an appended extension with antagonistic properties to inhibit LDLR binding. The extension must encroach upon the LDLR binding site’s hydrophobic pocket which significantly contributes to the binding energy of LDLR. Using two known PDB structures containing the removable peptides (A (5VLP) and B (6U3I)) in the N terminal grove, a three-dimensional printed model was created to demonstrate the interactions and proximity to the hydrophobic pocket. This model highlights critical amino acids on the peptides and PCSK9 to emphasize how the interactions support certain substitutions. One significant interaction lies between PCSK9 residues Ile369, Phe379, Asp238, and Ala239 and Peptide B’s organic moiety (1-amino-phenylcyclohexane-1-carbonyl). These PCSK9 residues surround the LDLR binding region's hydrophobic pocket indicating a successful inhibition. On the other hand, Peptide A’s FPG motif added to the Trp1 anchor formed a beta-turn which was unable to reach and, therefore, interact with PCSK9 residues Ile369, Phe379, Asp238, and Ala239. Through the 3D model, it was visualized that Peptide A had a beta-turn that precluded further extension into the target site, limiting its antagonistic ability. The model also highlighted Peptide B’s attached organic moiety which reaches the hydrophobic pocket in the proximal LDLR binding region, as shown in the literature to increase the binding affinity by >100 fold with reduced overall mass for an improved oral therapeutic.
Recommended Citation
Soni, Bhavya and Vemulapalli, Pritika, "Exploring structural differences between antagonistic peptides for the development of orally bioavailable PCSK9 inhibitors" (2022). Protein Modeling Reports. 8.
https://nsuworks.nova.edu/protein_modeling_reports/8
Poster
Presentation_PCSK9 Inhibitors.pptx (2298 kB)
Presentation
6U3I.jpg (338 kB)
Jmol Image of PDB 6U3I
5VLP.jpg (269 kB)
Jmol Image of PDB 5VLP